Votre panier est actuellement vide !
Compétences
Analyse de protéines
La précision, la sensibilité et la rapidité des spectromètres de masse modernes ont constitué une percée dans le domaine de la protéomique. Chez Allumiqs, notre équipe d’experts effectuera une gamme complète d’analyses allant de l’identification et de la caractérisation à la cartographie et à la quantification et fournira à nos clients des données significatives.
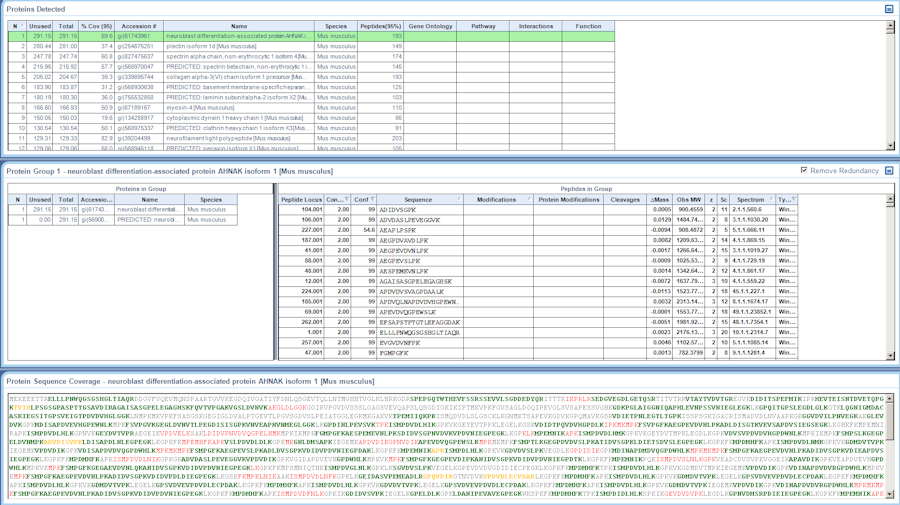
Identification des protéines
Identifier votre protéine d’intérêt par LC-MS/MS
Nous avons optimisé notre workflow pour l’identification des protéines afin de fournir des solutions rapides et efficaces compatibles avec la digestion en gel des protéines colorées au nitrate d’argent ou au bleu de Coomassie, ainsi que pour les protéines en solution.
Workflow d’identification des protéines sur gel
Préparation d’échantillons –>
Tout d’abord, nous lavons les morceaux de gel avec des cycles successifs de déshydratation/réhydratation dans des solutions compatibles avec la spectrométrie de masse. Dans une deuxième étape, nous rompons les ponts S-S intra- et inter-protéines à l’aide d’un agent réducteur et bloquons les cystéines libres par alkylation. Les protéines réduites sont ensuite digérées directement dans le gel à l’aide d’une protéase qui clive les résidus connus. Après la digestion, nous extrayons les peptides résultants du gel par des cycles successifs de déshydratation et de sonication. Enfin, ces peptides sont purifiés à l’aide d’une SPE (extraction en phase solide) en phase inversée et injectés dans le spectromètre de masse.
Analyse LC-MS/MS –>
Avant d’être analysés par spectrométrie de masse, les peptides sont séparés à l’aide d’un système de chromatographie liquide en phase inversée directement relié au spectromètre de masse. Une séparation supplémentaire permet une analyse approfondie des échantillons complexes et augmente la précision quantitative et qualitative. Une fois dans le spectromètre de masse, les peptides sont fragmentés et les signaux sont enregistrés par l’instrument à l’aide d’une méthode d’acquisition appelée acquisition dépendante des données (DDA), également connue sous le nom de mode d’acquisition dépendant de l’information (IDA). Le processus produit une liste de masses de précurseurs (MS) et de masses de fragments (MS/MS) en vue d’une analyse ultérieure.
Analyse des données et Identification des protéines
Pour obtenir l’identité des protéines présentes dans l’échantillon, nous chargeons les données dans un logiciel d’identification des protéines. Ce logiciel associe la liste des masses enregistrées à une liste de peptides et les attribue à une ou plusieurs protéines. Il convient de noter que ce processus signale également toute modification post-translationnelle, modification chimique ou substitution d’acide aminé susceptible d’être enregistrée.
Cartographie des peptides
Décoder les empreintes de protéines
La cartographie des peptides est utilisée pour confirmer la séquence d’une protéine. Après digestion par une protéase, les peptides sont séparés sur un système de chromatographie liquide et analysés par spectrométrie de masse. La protéase de choix pour réaliser la cartographie peptidique est la trypsine, car son activité est hautement prévisible et reproductible.
La cartographie des peptides est utilisée pour confirmer la séquence d’une protéine. Après digestion par une protéase, les peptides sont séparés sur un système de chromatographie liquide et analysés par spectrométrie de masse. La protéase de choix pour réaliser la cartographie peptidique est la trypsine, car son activité est hautement prévisible et reproductible.

Workflow de cartographie des peptides
Digestion In silico
La première étape de la confirmation de la séquence complète des acides aminés d’une protéine par spectrométrie de masse consiste à la découper en peptides plus petits à l’aide d’une protéase. L’enzyme de choix pour de nombreux workflows protéomiques est la trypsine, car elle coupe de manière fiable chaque résidu de lysine et d’arginine (à quelques exceptions près). Cependant, pour certaines parties de votre protéine, une digestion tryptique peut produire des peptides trop petits ou trop grands pour être correctement détectés par la machine. Il en résulterait un manque d’informations sur la séquence de ces parties, ce qui n’est pas souhaitable pour une expérience de cartographie peptidique. Pour éviter cela, nos experts procéderont d’abord à une digestion in silico de votre protéine d’intérêt afin de déterminer quelle combinaison de protéases produirait des peptides qui, une fois fusionnés de manière bio-informatique, couvriraient 100 % de la séquence. Nous obtenons généralement une couverture complète avec une combinaison de deux ou trois protéases différentes.
Digestion et LC-MS/MS
Une fois le choix des protéases effectué, nous réaliserons la digestion réelle de votre protéine en laboratoire. Les peptides résultant de chaque digestion seront analysés sur un gradient LC-MS/MS court en utilisant un workflow d’acquisition dépendant des données.
Analyse de données
Un logiciel d’identification des protéines associera d’abord les données enregistrées à une séquence peptidique pour chaque protéase. Il en résultera une liste de zones couvertes par des peptides dans chaque protéase individuelle. Enfin, nous regroupons et alignons chaque peptide confirmé de chaque digestion sur la séquence théorique de la protéine pour obtenir la cartographie peptidique finale.
Nos compétences en matière d’omique
Protéomique
Obtenez des données quantitatives pour jusqu’à 6500 protéines dans vos échantillons grâce à notre workflow de protéomique quantitative sans étiquette.
Lipidomique
Obtenez un profilage approfondi de nombreuses classes de lipides biologiquement importants grâce à nos instruments à haute résolution.
Métabolomique
La meilleure technique consiste à établir le profil de l’échantillon en utilisant la métabolomique non ciblée ou des panels sélectionnés de métabolites ciblés.
Analyse de données
Nous aidons nos clients à exploiter la valeur et le potentiel de leurs données en leur fournissant des informations plus claires et plus approfondies.
Entrez en contact
Communiquez avec nos experts